| |
Regulatory Process Overview: From Conception to Birth of an Idea
The cost of bringing a new drug to market has soared to between $800,000 and $1.7 billion, and can take as long as 15 years, according to the FDA. The increasingly challenging, inefficient, and costly medical product development path is keeping newer basic science discoveries from yielding more effective, affordable, and safer medical products for patients. During the last several years, the number of new drug and biologic applications submitted to the FDA has declined significantly; while the costs and difficulties of medical product development have soared.
The three-phase drug development process — ensuring safety, demonstrating efficacy, and manufacturing effective treatments — has changed little in the last 30 years. In many cases, developers must use the tools and concepts of the last century to assess this century's candidates. This results in failure for the vast majority of investigational products that enter clinical trials fail, but only after extensive investment of time and resources. This high failure rate drives up costs, forcing developers to use the profits from a decreasing number of successful products to subsidize a growing number of expensive failures. The path to market, even for successful candidates, is long, costly, and inefficient, due in large part to the current reliance on cumbersome assessment methods.
The FDA’s expedited development and review programs speed the availability of those drugs that promise significant benefits over existing treatments for serious or life-threatening diseases for which no adequate therapies exist.
Three-Phase process
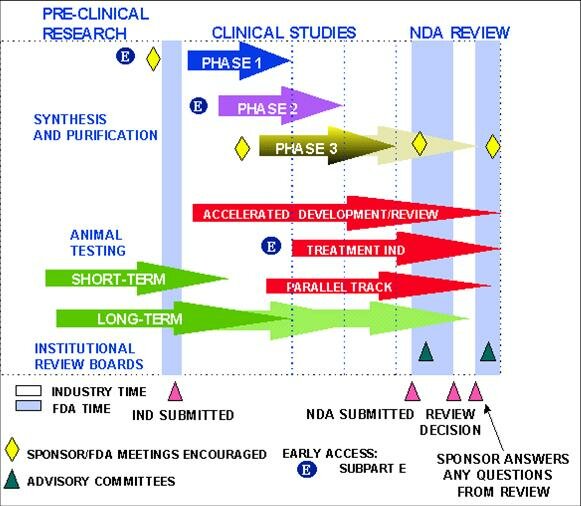
Pre-Clinical research and animal testing
The drug development process starts with a sponsor, usually a pharmaceutical company, seeking to develop a new compound it hopes will find a useful and profitable place in the market. Before an investigational drug can be tested in humans, researchers must prove that the compound is safe for use in small-scale, human clinical studies by first analyzing its physical and chemical properties in the laboratory and assessing its safety and therapeutic efficacy in animals.
Animal testing reveals what, if any, affect the drug has on a disease or its symptoms and at what dosage levels; the rate of absorption, metabolism, and elimination; and the drug’s therapeutic effect. Generally, two or more species of animals are tested because a drug may affect one species differently from another. Testing ranges from a few weeks to several years. Some animal testing even continues after human tests have begun in order to evaluate whether long-term use of the drug may cause cancer or birth defects. Since animals have a much shorter lifespan than humans, valuable information can be gained about a drug’s effects over an animal’s life cycle.
If the results of laboratory and animal study are promising, the treatment sponsor can file an Investigational New Drug application asking the FDA to approve human clinical trials.
Investigational New Drug (IND) application
The Investigational New Drug (IND) application includes:
-
Pre-clinical study data showing the product will not expose participants in limited, early-stage clinical studies to unreasonable risks.
-
An explanation of the drug’s intended medical use.
-
Proposed clinical trial protocols (e.g. how many patients will be involved, how will the drug be administered and at what dosages, names and qualifications of clinical investigators, and study locations).
For every 100 compounds for which IND applications are submitted to the FDA, about 70 percent will successfully complete phase I clinical trials and go on to phase II; about 33 percent of the original 100 will complete phase II and advance to phase III; and 25 to 30 of the original 100 will clear phase III. On average, about 20 of the original 100 compounds will ultimately be approved for marketing. (Tufts)
The FDA has 30 days to review an IND and approve or place it on “clinical hold” until issues are resolved. “Compassionate INDs,” allow patients with life-threatening diseases access to promising experimental drugs without being part of a clinical trial.
Pipeline Project Patient Advisors will play a vital role in this phase of treatment development.
Clinical trials: Protocol and Institutional Review Boards
The FDA relies on clinical trial results as a basis for concluding that a new drug has or has not shown “substantial evidence of effectiveness, as well as confirmation of relative safety in terms of the risk-to-benefit ratio for the disease that is to be treated. ”All clinical trials are based on protocol designed to answer specific research questions. The document must include eligibility criteria for participants; a schedule of tests, procedures, medications, and dosages; and the length of the study.
The FDA and a local Institutional Review Board (IRB) — an independent committee of physicians, scientists, statisticians, ethicists, and community advocates — must approve the protocol before recruitment can begin and must make sure participants are fully informed and willing, and have given their written consent before the trial is begun. In the process, IRBs:
-
Evaluate ethical aspects of the proposed study.
-
Ascertain a proposal’s acceptability in terms of institutional commitments and regulations, applicable law, standards of professional conduct and practice, and community attitudes.
-
Approve, modify, or disapprove research protocols.
-
Ensure the rights and safety of clinical trial participants.
-
Review the research as it goes along.
The research staff regularly monitors participants’ health to determine the safety and effectiveness of their treatment. The FDA Center for Drug Evaluation and Research (CDER) oversees clinical trials; conducts ongoing reviews of emerging data on safety, efficacy, and product quality; and engages outside experts to help review results. Should serious problems emerge, CDER informs the scientific community, and conducts or collaborates on relevant research. CDER can halt trials it deems unsafe or unlikely to meet stated objectives.
The Pipeline Project’s Research Partners/Patient Advocates will serve as ombudsmen on the front line in key clinical trial settings, facilitating the research and development process for new treatments. They will:
Put the face of the patient, and the urgency felt by all PWP, in front of our various constituencies.
Contribute the unique perspective and credibility of PWP.
Inform doctors about referrals to research programs.
Clinical trial phases
Clinical trials often take many years to complete. They are conducted in three main phases, each having a different purpose and helping scientists answer different questions. Each phase includes more participants than the previous one because investigational drugs that may appear to be safe and effective in initial testing, often reveal unanticipated, potentially serious side effects when tested in larger patient populations. Also, compounds that look effective based on preliminary data sometimes are shown to have little or no therapeutic benefit in later studies.
Phase I trials
-
Involve 20 – 80 healthy volunteers.
-
Assess safety over a period of six months to a year.
-
Establish a safe dosing for future trials.
-
Identify common, acute, adverse side effects.
-
Begin to clarify what happens to the drug in the human body (e.g. is the drug changed, how much of it gets into the blood and various organs, how long does it stays in the body, and how does the body gets rid of the drug and its effects).
The FDA reviews trial results and, if it concludes the drug is safe and may be beneficial against a particular disease or condition, testing can move to Phase II.
Phase II trials
-
Involve up to 300 people from selected patient populations who have the disease/condition to be treated or prevented.
-
Assess effectiveness and short-term safety over period of several months to two years.
-
Determine the common short-term side effects and risks associated with the drug.
-
Assign dose and dosing regimens for the magnitude and duration of effect.
Phase III trials
-
Involve 1,000 to 3,000 patients for one to four years.
-
Use randomized, controlled, double-blind studies, which have some participants receiving the active drug and others a different treatment or a placebo. Neither the investigators nor the patients know who is receiving what.
-
Confirm a drug’s safety and effectiveness, both clinically and in improving participants’ activities of daily living.
-
Determine at what doses the drug works best, and monitor side effects.
-
Compare the drug to commonly used treatments.
-
Define the drug’s overall benefit-to-risk ratio.
-
Collect information that will allow the drug or treatment to be used safely (e.g. does it interact with specific foods and/or other medications? Should certain patient populations avoid its use?) and include this information on the label.
Most drugs that reach this phase will be considered for approval by an FDA advisory board.
Phase IV trials
-
Conduct after a drug has been approved for marketing.
-
Identify drug’s risks, benefits, and optimal use.
-
Gather additional safety information from a larger group of patients.
-
Establish effectiveness in a subgroup of patients (e.g. patients over age 65) not previously studied.
-
Determine incidence of adverse reactions and long-term effects.
-
Make marketing comparisons against other products and other uses.
New Drug Application (NDA)
Once a new drug is deemed relatively safe and effective, the sponsor can submit a New Drug Application (NDA) to the FDA, seeking permission to manufacture and market the drug. The NDA must include all data from animal and laboratory testing, information about the drug’s chemistry and pharmacology, and results of clinical investigations. The FDA examines the drug’s safety and efficacy data, analyzes drug samples, checks product labeling for scientific accuracy. This review process can take months to several years, depending on the complexity of the application.
Sponsors need not prove that a new drug will be 100% safe and effective for all patients, as there is always some risk of potential adverse reactions when using prescription drugs. The FDA's approval decisions, therefore, always involve an assessment of the benefits and the risks for a particular product. A drug with a higher risk profile may be approved if it is believed to have benefit for patients with a serious or life-threatening disease, and there is no alternative therapy available. The FDA may insist additional studies or data analysis be conducted before the application is considered for approval. When the benefits of a drug are thought to outweigh the risks, and if the labeling instructions allow for safe and effective use, the FDA considers a drug safe for approval and marketing. The FDA has 180 days to either approve or disapprove an NDA for serious deficiencies (e.g. inadequate tests).
Postmarketing surveillance
The FDA’s MedWatch program conducts “postmarketing surveillance” of medical products to identify safety concerns that appear after a drug is marketed and determine whether these adverse effects are serious enough to warrant regulatory action. Where side effects pose an “imminent hazard” to public health, the FDA can order the drug to be immediately withdrawn from the market. More typically, the agency will require labeling changes, and issue a general warning through the media, its website, and “medical alert” letters to health professionals.
The MedWatch program’s goals are to:
-
Clarify what should and should not be reported to the FDA.
-
Increase awareness of serious reactions caused by drugs or medical devices.
-
Make the reporting process easy.
-
Give the health community regular feedback about product safety issues.
Health professionals and consumers are asked to submit a MedWatch report if there is a suspected causal relationship between the use of a drug and a serious adverse reaction — one that resulted in death, life-threatening hazard, hospitalization, medical or surgical treatment to prevent impairment, disability, and birth defects, miscarriage or still birth. The FDA analyzes the magnitude of the health risks involved, determines if the drug in question is actually responsible, and decides whether these factors justify remedial action.
Resources and links:
FDA: "From Test Tube to Patient: Improving Health Through Human Drugs"
(a pdf file)
ClinicalTrials.gov: Introduction to Clinical Trials
|